RESEARCH研究内容
遠紫外分光法
凝集相における遠紫外分光法の創成―新しいσ電子化学を目指して
遠紫外領域には数々の電子許容遷移が観測される。しかし遠紫外域ではモル吸光係数が非常に大きいため、これまで大方、気相でスペクトル測定が行われてきた。凝集相にとっては分子分光学の未踏領域となっていた(1-5)。尾崎教授らはこの未踏領域の開拓に挑戦した。
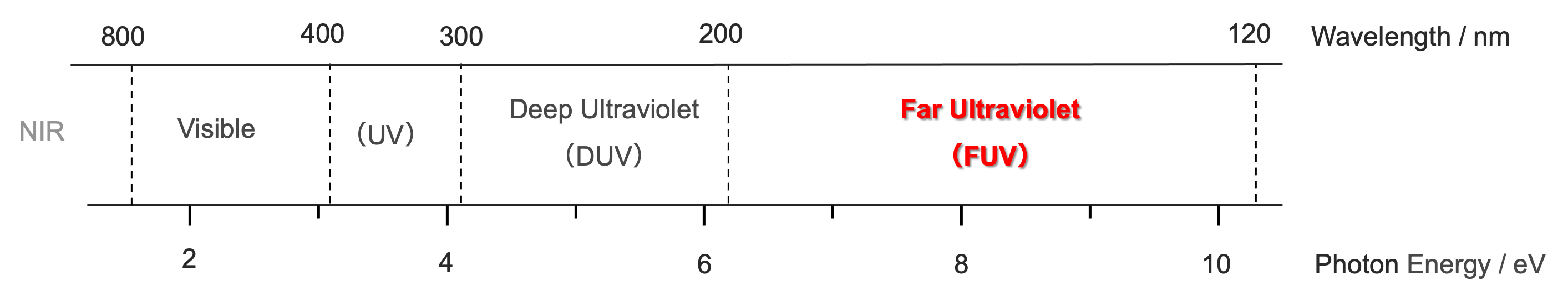
図1 可視から遠紫外までの領域
ここでは,紫外領域を紫外(400-300 nm),深紫外(300-200 nm),遠紫外(200-120 nm),真空紫外(極端紫外)(120-10 nm)に分ける(1-5).遠紫外領域では分光器を真空に引く,または窒素ないしアルゴンガスで置換する必要があるが,深紫外領域ではそれらの必要がない.したがって二つの領域の間にははっきりとした境が成立する.しかし,遠紫外域は必ずしも真空を必要としないので,この領域を真空紫外域に入れるのは適切ではない.図 1 から明らかなように,遠紫外域はエネルギーから考えるとかなり広い領域である.
遠紫外域では,溶媒も含めてすべての分子が許容電子遷移を持ち,モル吸光係数が非常に大きくなるので(εFUV=104-106 mol-1 dm3 cm-1),これまで主に気体を対象として研究が進められてきた(1-5).一方、遠紫外域での凝縮相の吸収スペクトル測定は,吸光度が非常に大きいこと,目立った応用が考えにくいことなどの理由から、あまり行われてこなかった.そこで吸光度の問題を解決するために、尾崎教授ら(池羽田、東)は、遠紫外域に ATR 法を導入した。(6). ATR 法の導入により,これまで測定が不可能であった水,アルカン,アルコール,アミドなどほとんどすべての分子の凝縮相での遠紫外スペクトル測定が可能になった(1-5).ATR-遠紫外分光法の確立によって電子分光学の世界が大きく拡がり,固体や液体の電子遷移や電子状態の研究が大きく進展した.とくに遠紫外分光法と量子化学計算法を組み合わせた研究は,測定されたスペクトルのバンドの帰属を可能とするだけではなく,電子遷移や電子状態の研究さらに分子間相互作用の研究などを発展させた(1-5,7-10).図2にATR-遠紫外分光装置の外観と装置図を示す。この装置の詳しい内容は文献2, 6aで解説した.この装置では遠紫外部である程度透明でありながら,高い屈折率を持つサファイヤを内部反射素子(IRE)として用いている.この装置の利点は,分光器内部は窒素ガスでパージする仕組みになっているが,試料部は空気中にあるため,試料の交換が容易で,試料に対するワークスペースを十分に取れるというという点である.
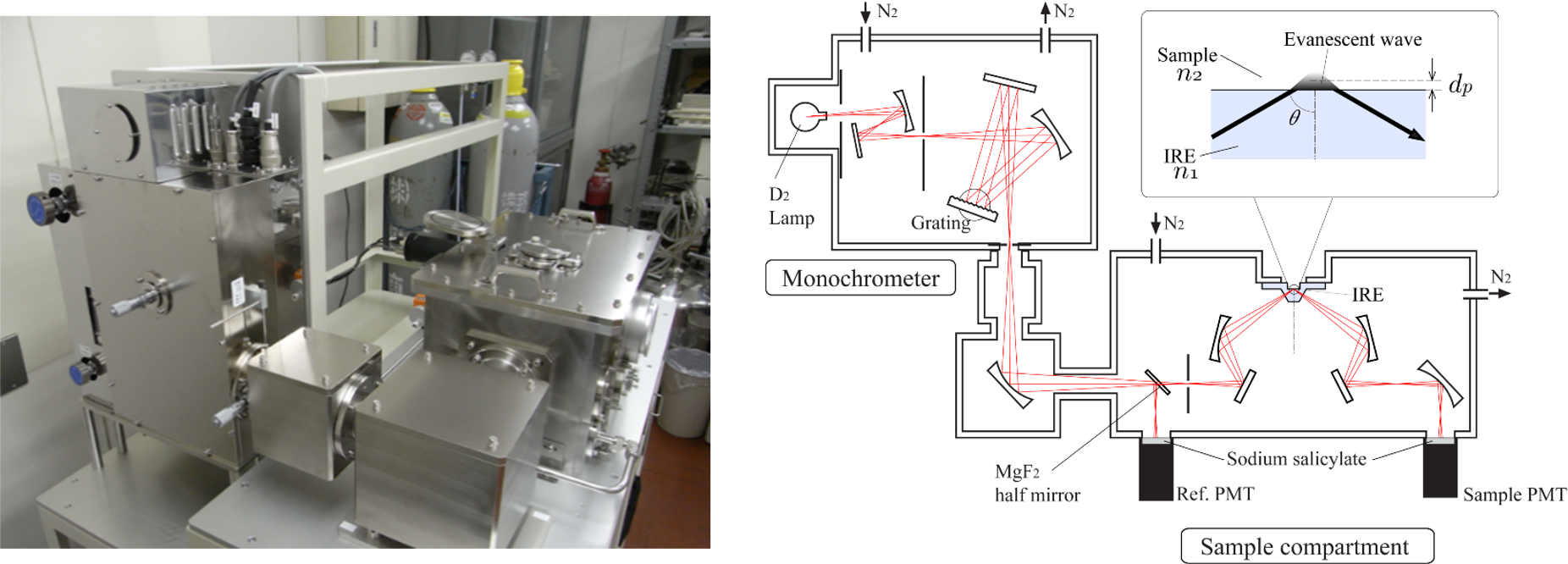
図2 尾崎研で東、池羽田らが開発したATR-遠紫外分光器の外観と装置図
遠紫外分光法の特徴と利点
ATR-遠紫外分光法の特徴と利点は以下のようにまとめることができる(1-5).
1. 遠紫外分光法は分子の電子状態や電子遷移について通常の紫外分光法では得られない情報を与える.たとえば Rydberg 遷移やσ電子の関係する遷移などである.
2. 水,アルカン,アルコールなど通常の紫外領域に吸収を示さない分子が遠紫外域にはバンドを示すので,これらの物質の電子状態や分子構造などを研究することが可能である.
3. 水素結合,分子内,分子間相互作用,水和,相転移などについてユニークな情報を与える.
4. ATR 法を用いることにより,~100 nm 付近の表面,界面における電子状態,分子構造の研究が可能となる.多角入射ATR法を用いると界面深さ依存性について調べることができる.
5. シンクロトロン放射に基づく遠紫外分光装置に比べ, ATR-遠紫外分光装置ははるかに小型で, 操作が容易で, 費用が安くつく.
6. 時間分解ATR-遠紫外分光法を用いることにより、光化学反応のキネティックスの研究を行うことができる。
7. ATR-遠紫外分光法と紫外励起共鳴ラマン分光法を組み合わせることにより、例えば、タンパク質やDNAの電子状態と分子構造の関係を調べることができる。
これまで遠紫外分光法は,基礎研究としては有機分子を中心とした分子の電子状態,電子遷移の研究に用いられている.中でも注目されるのは,凝縮相における Rydberg 軌道が関与する電子状態の実験的,理論的研究や(7-9),分子間相互作用が分子の単結合骨格の価電子(σ電子)に与える影響の研究などである(10).
遠紫外分光法の応用は以下にまとめるように,きわめて広範囲に及んでいる(1-5).
1. 水,水溶液の研究(水の電子状態,カチオン,アニオンの水分子の電子遷移への影響の研究,表面吸着水の研究など)(6,11,12)
2. 材料科学への応用.ポリマー(13-16),TiO2をはじめとする無機半導体材料(17,18),イオン液体(19),グラフェンなどのナノカーボン材料(20),ポリマーナノコンポジットなどの電子状態の研究(15),TiO2 の光触媒活性の研究(17,18)
3. 生命科学への応用
4. 電気化学への応用(21)
5. ATR を利用した表面,界面,超薄膜の研究.多角入射 ATR 法の利用(12)
6. 時間分解遠紫外分光法によるラジカル反応や光化学反応の研究(22)
7. 遠紫外領域における表面プラズモンの研究とその応用(23)
8. プロセスモニタリング,地質分析,環境科学への応用(24,25)
有機化合物の電子遷移,電子状態の研究
森澤ら(7-9)は ATR-遠紫外分光法を用いてアルカン、アルコール、ケトンの研究を系統的に行った.彼らの研究で最も重要な成果は,凝縮相においても Rydberg 遷移が存在することを立証したことである.アルカンやケトンの遠紫外スペクトルには,Rydberg 遷移を含めて考えなければ,説明できないバンドが観測された.量子化学計算でRydberg 遷移を取り扱う場合は,時間依存密度汎関数法(Time-dependent density function theory, TD-DFT)では汎関数には CAM-B3LYP 法を,基底関数には分散関数を加えることでよい結果が得られた.アルカンに関しては波動関数理論である SAC-CI (Symmetry adapted cluster/configuration interaction)法を用いても Rydberg 遷移の存在を確認できた(7-9).
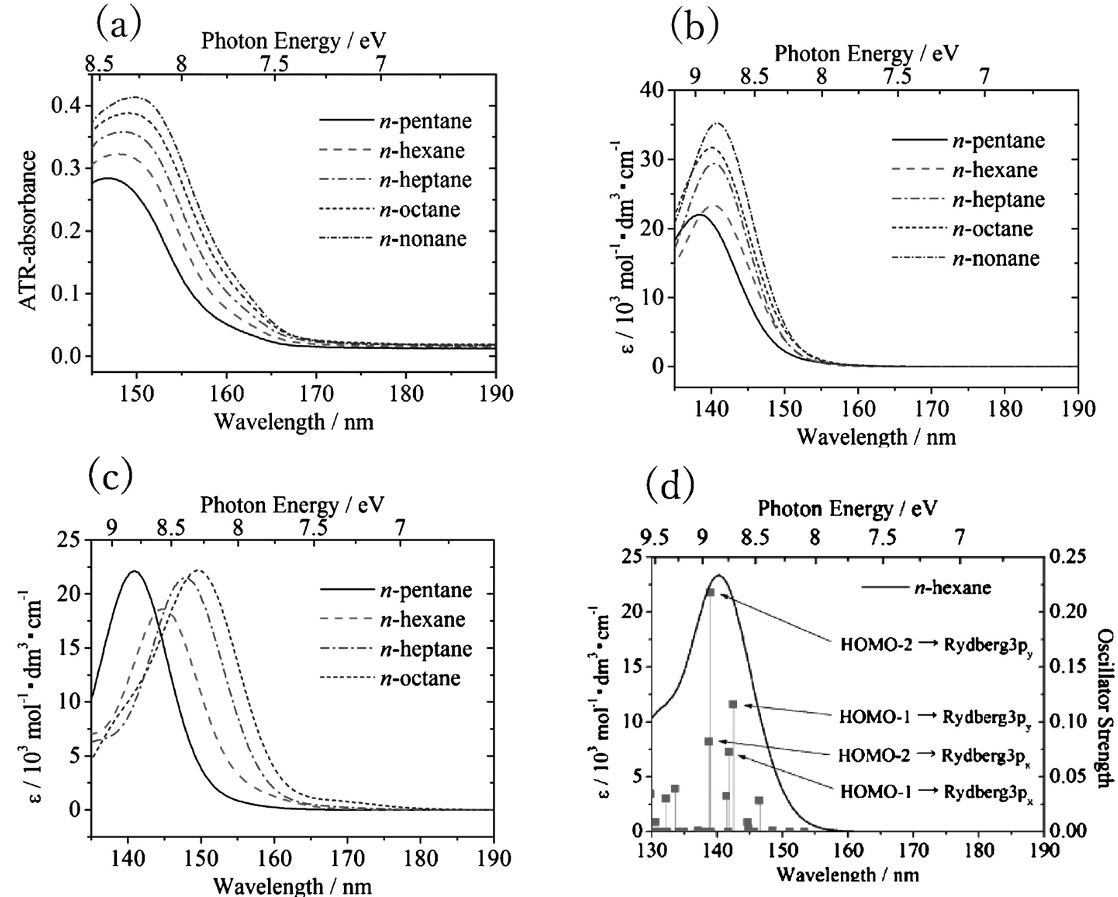
図 3(a) n-アルカン(CnH2n+2, n=5-9)の ATR-遠紫外スペクトル.(b), (c)CAM-B3LYP 法と SAC-CI 法(いずれも基底関数には aug-cc-pVTZを用いた)を用いた量子化学計算の結果. (d)n-ヘキサンのバンドの帰属(8).
図 3(a)は n-アルカン(CnH2n+2, n=5-9)の ATR-遠紫外スペクトルである(8).いずれのアルカンも150 nm 付近に強い吸収を示すが,この吸収はアルキル鎖が長くなるにつれて長波長側にシフトし,しかも強度が強くなる.このスペクトルを解析するために,量子化学計算が行われた.CAM-B3LYP 法と SAC-CI 法(いずれも基底関数には aug-cc-pVTZを用いた)を用いた量子化学計算の結果をそれぞれ図 3(b), (c)に示す(8).いずれの量子化学計算の結果も,アルキル鎖の鎖長の増大に伴うピークの強度増大やシフトなどの点において実験スペクトルと良い一致を示したが,長波長シフトや遷移エネルギーの値については SAC-CI の方がよく一致している.一方,強度変化については CAM-B3LYP 法の方がよく一致した.図 3(d)に n-ヘキサンのバンドの帰属を示した(8).図3(d)で最も強い強度を与える遷移は,HOMO-2(σ)から Rydberg 3py への遷移(以下 T1 と名付ける)である.図 4 は量子化学計算によって得られた軌道の等電子密度面である(8).いずれのアルカンもヘキサンの T1 に相当する吸収ピークを与えるが, HOMO-2(σ)のエネルギー準位は鎖長とともに上昇し,n-へプタンで HOMO-3(π)と入れ替わる.HOMO-1(π)から Rydberg 3py への遷移(T2)は 2 番目に強いピークを与える.量子化学計算の結果から,アルキル鎖が長くなるとともに155 nm のピークが長波長シフトするのは,σ準位が不安定化するとともに,Rydberg 3p 準位が安定化するためと分かった(8).アルカンは単結合のみで形成される分子であり,非共有電子対を持たないことから,この遷移がアルカンのσ電子の状態を反映していることが明らかになった.
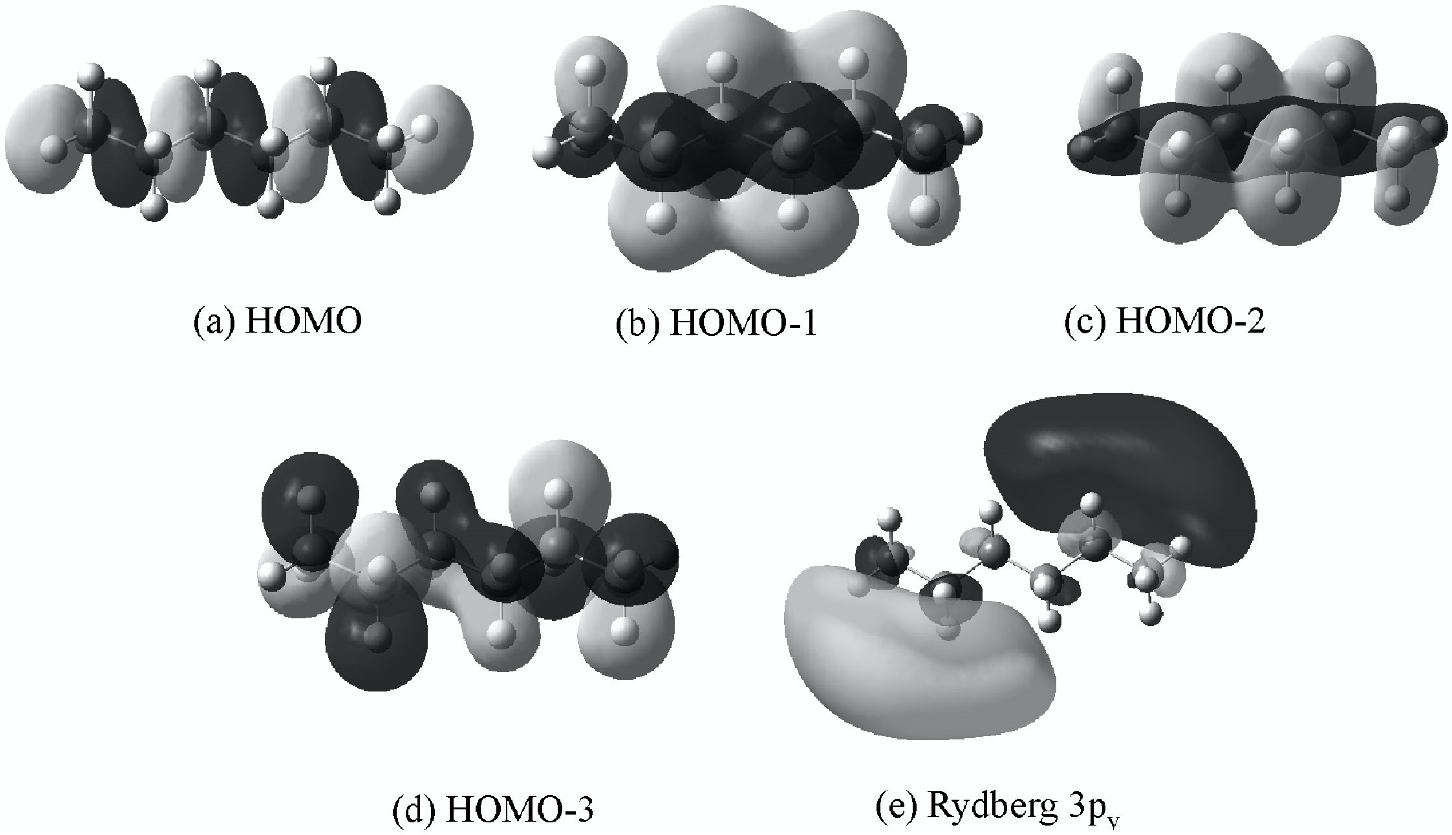
図4 n-hexaneのKohm-Sham 軌道; (a) HOMO, (b) HOMO-1, (c) HOMO-2, (d) HOMO-3, (e) Rydberg 3py (8).
森澤らはさらに遠紫外分光法を用いて、n-アルカンの分子間相互作用の研究を行った(10). 固体状態におけるアルカンの分子間相互作用を調べるために,常温で液体の n-テトラデカン(C14H30)を融点以下まで冷却したところ,相転移前後でスペクトル変化が観測された(10).153 nm のバンドの強度は融点(6°C)以下で単調に減少した.一方,200と230 nm に新しいバンドが融点付近で現れ,その後,単調に増大した.この温度変化は可逆的であった.この興味深い実験結果を解析するために,n-ペンタンダイマーのスペクトルの量子化学計算が行われた(10).ダイマー間の距離を 6 Å にとると,液体,すなわち常温の n-テトラデカンのスペクトルによく似たスペクトルが得られた.ダイマー間の距離を 3 Å まで縮めると固体状態のモデルとなり,-38°C の n-テトラデカンのスペクトルによく似たものが得られた(10).この実験結果と量子化学計算の結果から,森澤らはシフトの原因は,外側に張り出した σ軌道,HOMO-1 および HOMO-2 軌道が固体中の分子間相互作用により不安定化軌道と安定化軌道を作り,この二つの軌道間の遷移が低エネルギー側に観測されることによると結論した.このように遠紫外分光法でσ軌道の変化による遷移の変化を観測した例はこれまでにない.これは,まさに新しいσ電子化学を開拓する第一歩となる成果と言える(10).
アミドは, valence-Rydberg カップリングがπ-π* 遷移において期待されること,さらに水素結合による直接の相互作用が励起状態に影響するという点で興味深い(9).森澤ら(9)はスペクトル測定と量子化学計算の結果から,アミドの190 nm 付近のπ-π* 遷移によるバンドの周辺にいくつかの Rydberg 遷移が観察されること,π-π* 遷移と同じ対称性を持つ遷移のみが強度を持つことを示した.アミドの研究はナイロンの研究につながった(14).α-型ナイロン 6 やナイロン 6/6 など 5 種類のナイロンの ATR-遠紫外スペクトルが測定された.いずれのナイロンもアルカンによる吸収を 150 nm 付近に,アミドによる吸収を190 nm 付近に示すが,これらのバンドはナイロンの種類に依存するシフトを示した.ナイロンのスペクトルを解析するために,ナイロンのモデルを用いて量子化学計算が行われた.その結果,Rydberg 遷移はアルキル鎖に非局在化しているのに対し,π-π* 遷移はアミド基に比較的局在化していることが分かった(14).α-型のナイロン-6 とナイロン-6/6 の遠紫外スペクトルにおけるπ-π* 遷移によるバンドの波長を比較すると,後者の方が2nm長波長シフトする.これは結晶構造と分子間水素結合の違いによるものであることが明らかになった.これらの違いは,π-π* 遷移の局在化の違いや遷移双極子カップリングの違いとしても現れる(14).ナイロンの研究から、アルカン部分による吸収とアミド部分による吸収が特性吸収帯のように別々に独立して観測されることが分かった。
表1 縁紫外領域に観測される電子遷移 (森澤ら)
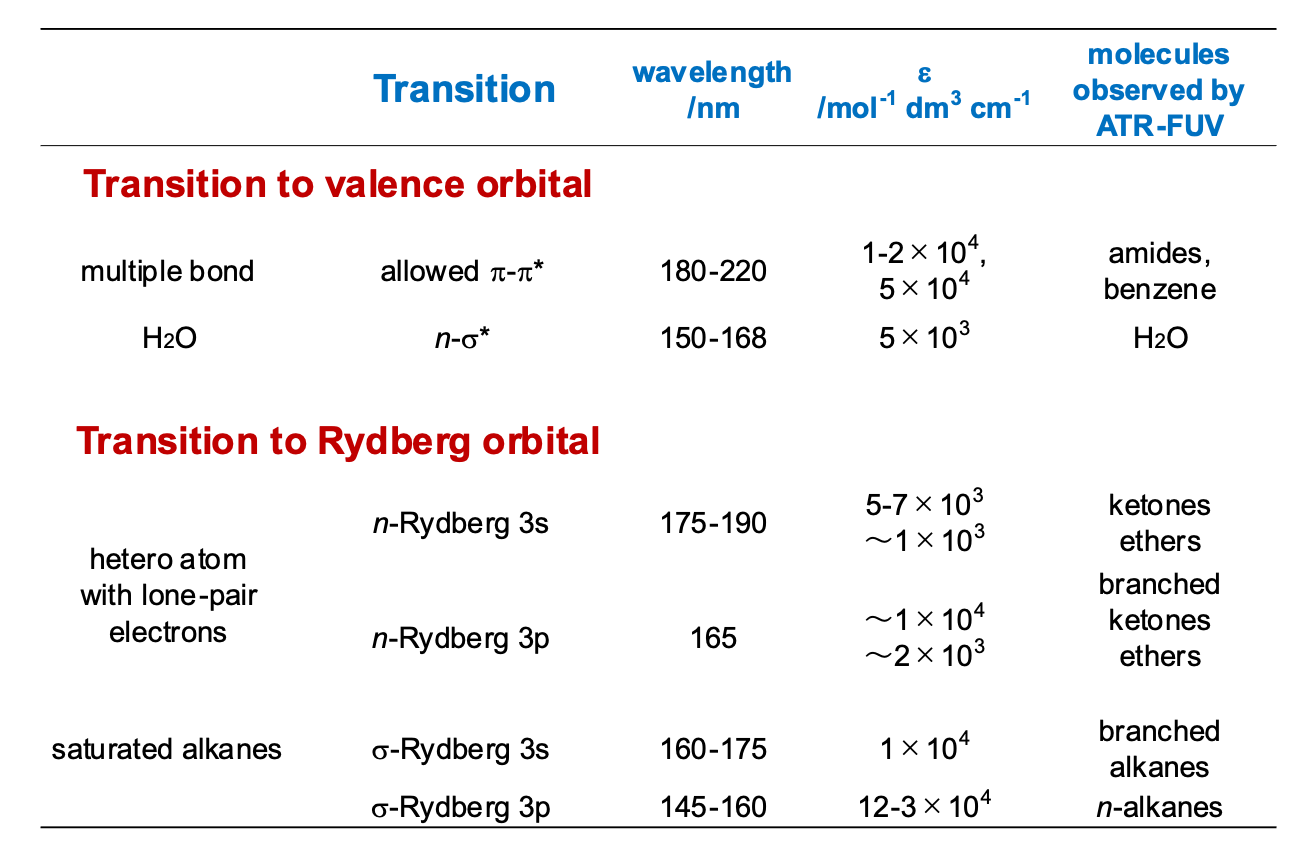
表 1 は ATR-遠紫外分光法と量子化学計算で明らかになった遠紫外域における遷移をまとめたものである.いずれの遷移も104-106 mol-1 dm3 cm-1 という大きなモル吸光係数を持つ.(1)許容π-π* 遷移―共役しない多重結合をもつ分子のπ-π* 遷移は180-220 nm に観測される.(2)n-σ* 遷移―O, N, S,ハロゲンを含んだ有機分子はn軌道からの遷移を示す.(3)Rydberg 遷移―二重結合を持った炭化水素ではπ-Rydberg 遷移が,飽和炭化水素ではσ-Rydberg 遷移が遠紫外域に観測される.
無機半導体粉末への応用
田邉らは酸化チタンをはじめとした無機半導体材料に ATR-遠紫外分光法を適用し,その電子励起スペクトルと光触媒活性との相関を明らかにした(17,18).図5a は表面に Pt, Pd, Au のナノ粒子(粒径数 nm)を修飾したアナターゼ型 TiO2 粉末の ATR-遠紫外スペクトルを示す(17).修飾金属の仕事関数が大きいほど ATR スペクトルが大きく変化しており(図5b),TiO2 の電子状態が変化していることが分かる.実際,材料の光触媒活性と修飾金属の仕事関数の間にも古くから知られているように正の相関がみられる(図5c).また,修飾金属依存性だけでなく,修飾金属のサイズや形状に対する依存性,TiO2 の結晶形やサイズ依存性についても,スペクトルと光触媒活性との関係が明らかになった(18a).
田邉らはまた,従来の ATR-遠紫外装置に測定試料への光照射機構を導入し,Au ナノ粒子修飾 TiO2 への光(紫外光と可視光)照射に伴うスペクトル変化を調べた(18).すなわち,紫外光照射に伴い TiO2 の ATR 強度が減少し(図6a),可視光照射に伴い増加する(図6b)という変化が測定された.これらの変化は紫外光照射に伴う TiO2 から Au への電子移動と,可視光照射に伴う Au から TiO2 への電子移動を反映すると考えられる(図7).
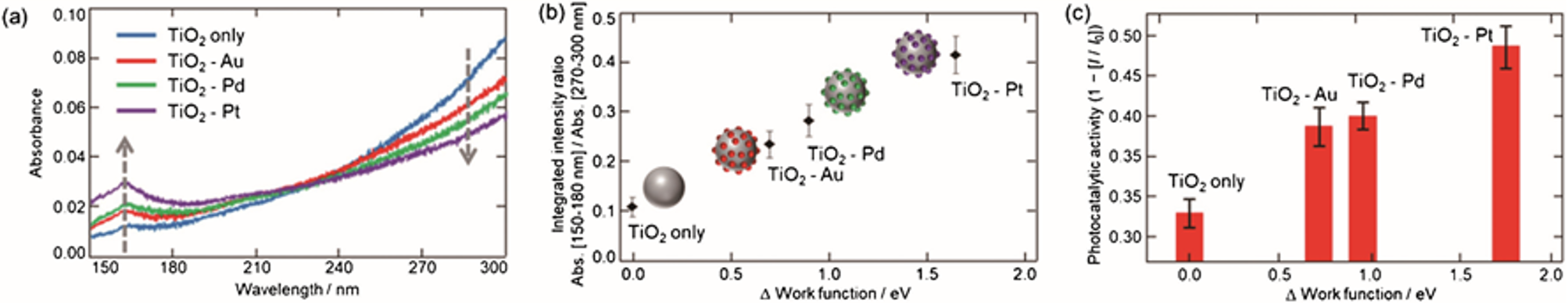
図5 (a) 表面に Pt, Pd, Au のナノ粒子(粒径数 nm)を修飾したアナターゼ型 TiO2 粉末の ATR-遠紫外スペクトル. (b) 150-180 nmと270-300 nmの領域の面積強度比とTiO2と各々の金属の仕事関数の差との関係. (c) 各々の材料の光触媒活性 (17a).
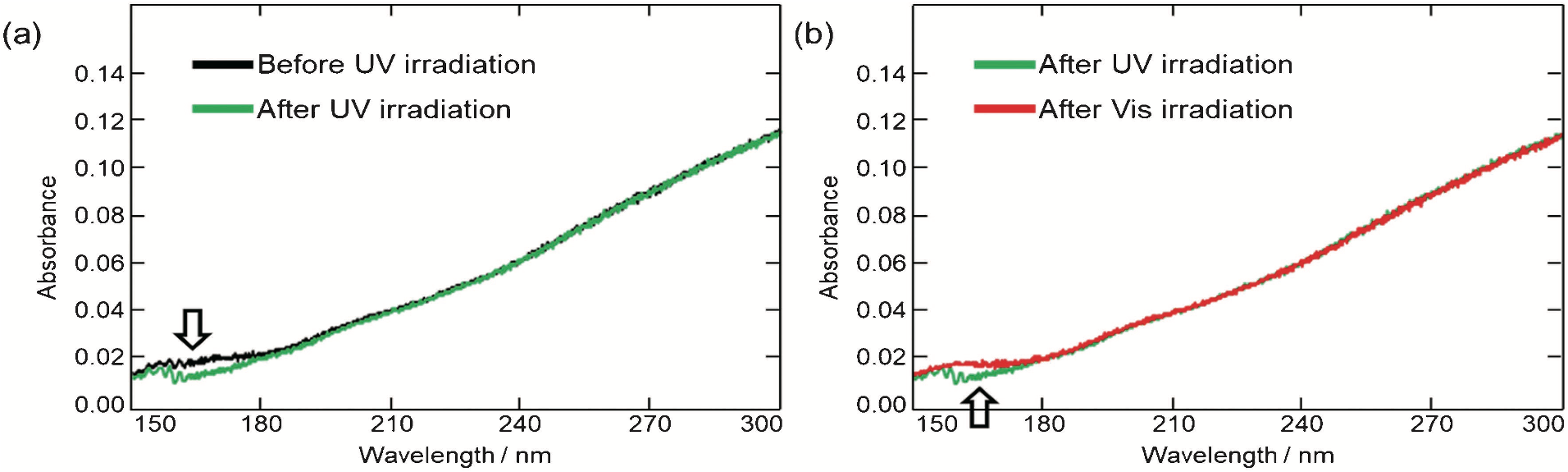
図6 (a) 紫外光照射前後のAu-TiO2の典型的な吸収スペクトル. (b) 紫外光照射と可視光照射の比較 (18b).
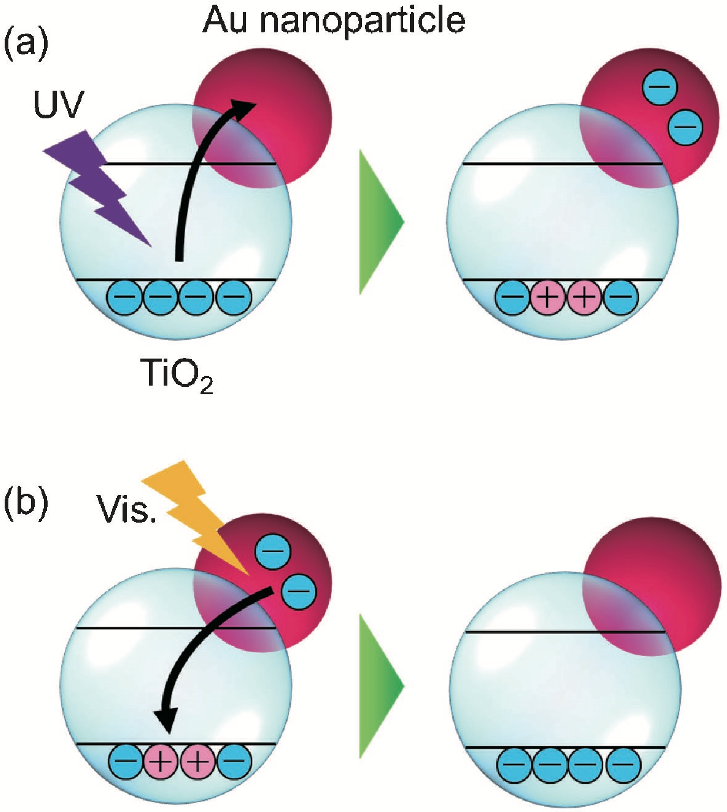
図7 電子移動の模式図. (a) 紫外光照射下におけるTiO2から金ナノ粒子への電子移動, (b) 可視光照射下における金ナノ粒子からTiO2への電子移動.
以上のように,ATR-遠紫外法は無機半導体材料の測定にも適用でき,さらにその材料機能評価手法としての有用性も示されつつある.特に,単結晶ではなく実用的に使われている粉末状態での測定が簡便にできることや,機能を発現する光照射中のスペクトルを測定できることは,本装置の大きな長所と言える.また田邉らは ATR-遠紫外法を新規電解質材料などへの応用も期待されるイオン液体にも適用した(19a). 最近では独自の電気化学環境下で測定可能なセットアップを構築し,イオン液体の ATR スペクトルの電位依存性も明らかにした(19b).無機半導体をはじめとした光機能性材料だけでなく,電気化学材料など幅広い機能性材料への ATR-遠紫外分光法の応用が期待される.
遠紫外,深紫外領域における表面プラズモン共鳴とその応用
田邉ら(23)は深紫外光, 深紫外光を利用した新しい表面プラズモン共鳴(Surface Plasmon Resonance; SPR)の基礎研究とそれに基づく SPR センサーの開発を進めている(23).遠紫外,深紫外光を用いることにより,以下の三つの利点が得られる.(1)遠紫外,深紫外域で物質が大きな誘電率を持つため,高いセンサー感度が期待できる.(2)短波長の光を利用するので,極表面の空間選択的センシングが実現できる.(3)遠紫外,深紫外域に各物質が固有の吸収を持つため,物質選択的センシングが可能である.特に,物質が数多くの共鳴を示す遠紫外域を利用することにより実現される物質選択性は,これまでの SPR センサーにはない全く新しい特色である.田邉ら(23)は遠紫外,深紫外光を用いた表面プラズモンの基礎研究として,プリズムの材質依存性,Al の膜厚依存性,測定試料の吸収の影響などについて調べた.
ここでは,Al 薄膜表面の雰囲気を空気から 1,1,1,3,3,3-ヘキサフルオロ-2-プロパノール(HFIP)に変化させた時の,SPR 特性の変化について述べる.Al 薄膜表面が空気のとき,入射角度を変えながらスペクトルを測定したところ,入射角度が大きくなるに従って SPR 波長が短波長側(高エネルギー側)にシフトする様子が観察された(図 8a).また,例えば波長200 nm での反射率の入射角度依存性を調べると,約55°に SPR に基づくディップが観察された(29).Al 薄膜表面に HFIP(屈折率~1.28)をキャストして同様の測定を行った結果,SPR 波長はレッドシフトし(入射角度75°では約190 nm→約280 nm),反射率角度依存性のディップは高角側に観察された(図8b).これらの変化は従来の可視 SPR センサーの変化量に匹敵し,酸化アルミの影響も考慮に入れたフレネルの式に基づくシミュレーション結果とも一致した.以上のように,Al 薄膜の遠紫外域における SPR 特性を測定し,さらに Al 表面の屈折率変化における SPR 特性変化の検出に成功した(23a).
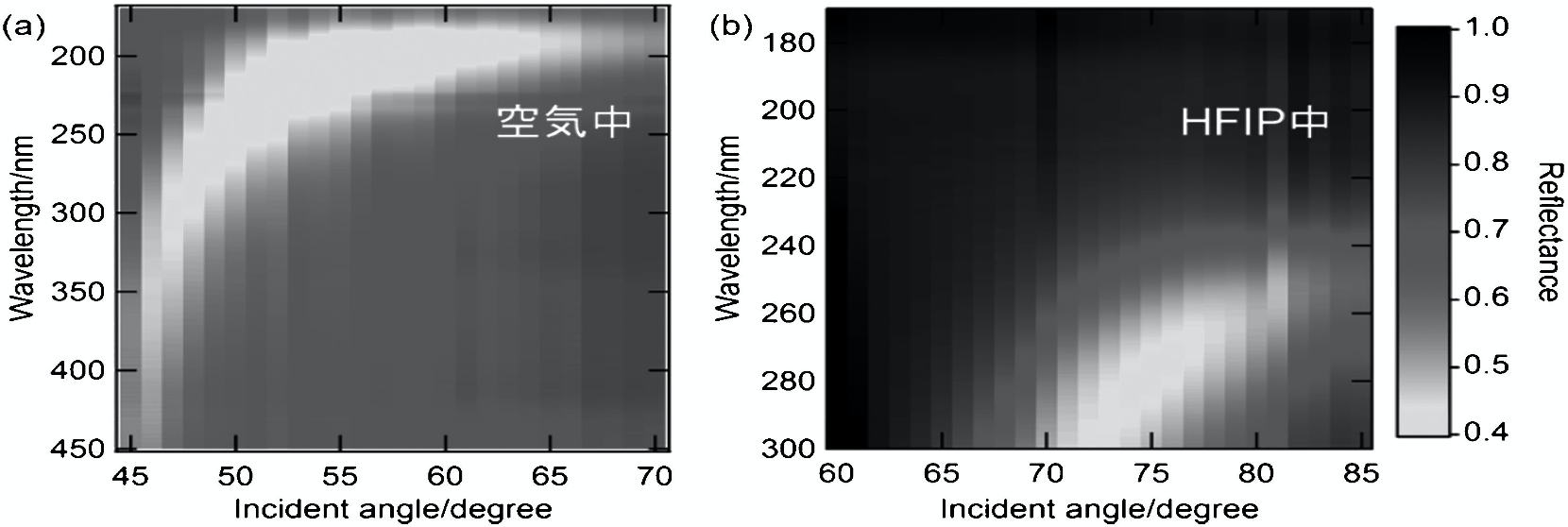
図8 (a)空気中と(b)HFIP中における反射率の入射角と波長依存性(23a)
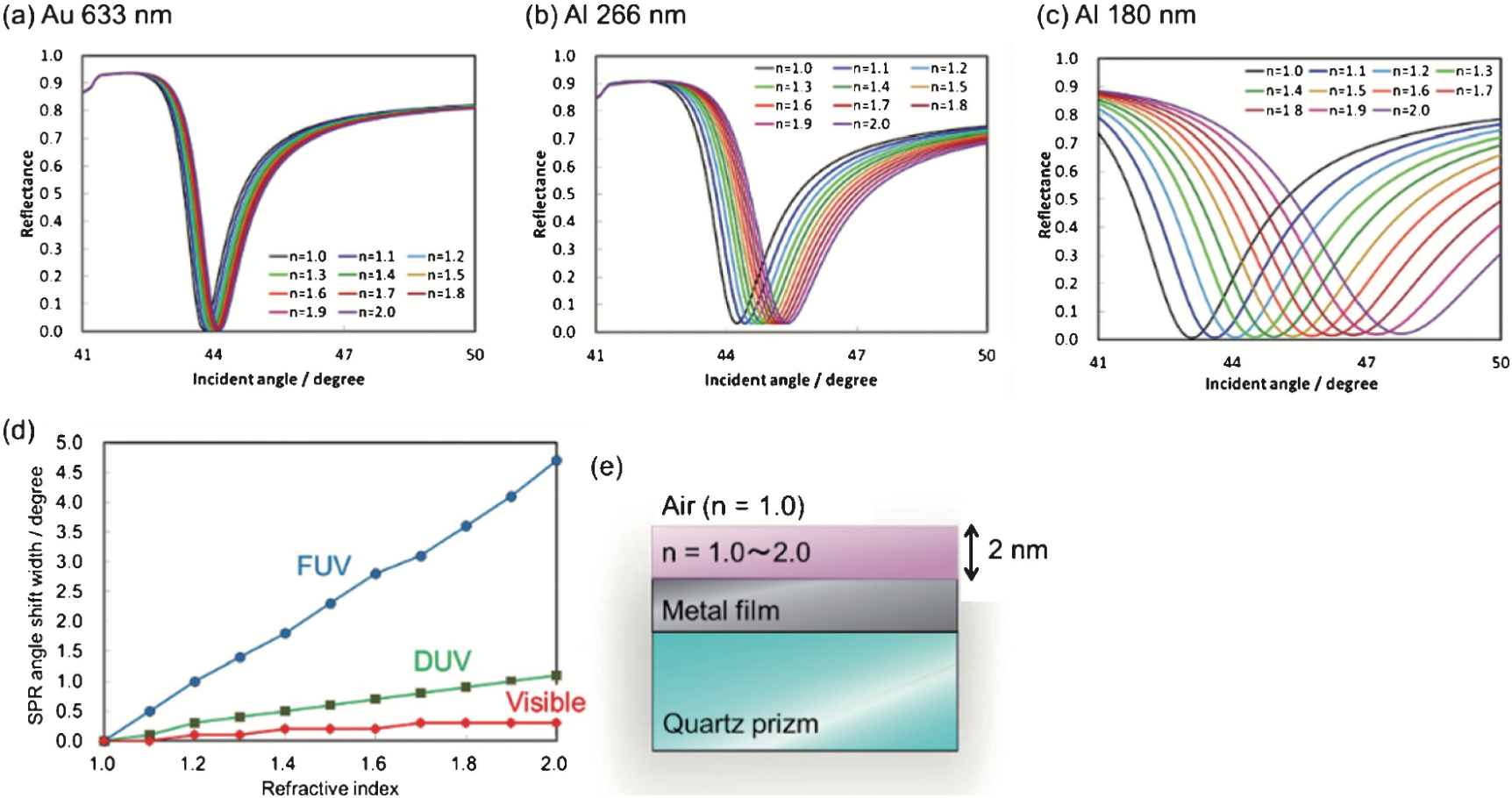
図9 周辺の反射率を変えた時の(a) 633nmにおける金フィルム,(b)266nmにおけるアルミフィルム,(c)180nmにおけるアルミフィルムの入射角依存性の計算結果.(d)SPR角の計算シフトの反射率依存性,(e)シムレーションのモデル(23b).
また本系では,従来の可視域よりも波長の短い光を利用するので,高い表面選択性が期待される(23).フレネルの式に基づくシミュレーションから,可視域に比較して遠紫外域は金属表面 1 nm の領域の屈折率変化を鋭敏に検出しうることが示された(図9).紫外域で各物質固有の電子励起に伴う屈折率の異常分散を利用した物質選択性と合わせて,新しい SPR センサーとしての応用が期待される.
参考文献
1) a) Y. Ozaki and S. Kawata, eds.: Far-and Deep-Ultraviolet Spectroscopy, Springer (2015). b) Y. Ozaki, Y. Saito, S. Kawata, in Ref. 1a. pp. 1-16.
2) a) 東昇,池羽田晶文,尾崎幸洋, 分光研究,57, 2-11 (2008).b) 森澤勇介, 分光研究、65, 91-103 (2016).c) Y. Ozaki, Y. Morisawa, A. Ikehata, and N. Higashi: Appl. Spectrosc., 66, 1-24 (2012).
3) Y. Morisawa, I. Tanabe, and Y. Ozaki: in “Frontiers and Advances in Molecular Spectroscopy”, J. Laane ed., Elsevier (2018). pp. 251.
4) a) Y. Ozaki and I. Tanabe: Analyst, 141, 3962 (2016). b) I. Tanabe, Y. Ozaki, J. Mater. Chem. C 4, 33, 7706 (2016).
5) Y. Ozaki, Y. Morisawa, I. Tanabe, and K. B. Bec, Spectrochim. Acta. A. 253, 119549 (2021).
6) a) N. Higashi, A. Ikehata, and Y. Ozaki: Rev. Sci. Instrum., 78, 103107 (2007). b) A. Ikehata, Y. Ozaki, and N. Higashi: J. Chem. Phys., 129, 234510 (2008).
7) a) Y. Morisawa, A. Ikehata, N. Higashi, and Y. Ozaki: Chem. Phys. Lett., 476, 205 (2009). b) Y. Morisawa, A. Ikehata, N. Higashi, and Y. Ozaki: J. Phys. Chem., 115, 562 (2011).
8) Y. Morisawa, S. Tachibana, M. Ehara, and Y. Ozaki: J. Phys. Chem., 116, 11957 (2012).
9) Y. Morisawa, M. Yasunaga, R. Fukuda, M. Ehara, and Y. Ozaki: J. Chem. Phys, 139, 154301 (2013).
10) Y. Morisawa, S. Tachibana, A. Ikehata, T. Yang, M. Ehara, and Y. Ozaki: ACS Omega, 2, 618 (2017).
11) a) A. Ikehata, M. Mitsuoka, Y. Morisawa, N. Kariyama, N. Higashi, and Y. Ozaki: J. Phys. Chem., 114, 8319 (2010). b) T. Goto, K. B. Bec , and Y. Ozaki: Phys. Chem. Chem. Phys. 19, 21490 (2017).
12) T. Goto, A. Ikehata, Y. Morisawa, and Y. Ozaki: J. Phys. Chem. Lett., 6, 1022 (2015).
13) H. Sato, N. Higashi, A. Ikehata, and Y. Ozaki: Appl. Spectrosc., 61, 780 (2007).
14) Y. Morisawa, M. Yasunaga, H. Sato, R. Fukuda, M. Ehara, and Y. Ozaki: J. Phys. Chem. B, 118, 11855 (2014).
15) K. B. Bec, Y. Morisawa, K. Kobashi, J. Grabska, I. Tanabe, E. Tanimura, H. Sato, M. J. Wojcik, and Y. Ozaki: Phys. Chem. Chem. Phys., 20, 8859 (2018).
16) N. Ueno, T. Wakabayashi, H. Sato, Y. Morisawa, J. Phys. Chem. A, 123, 10746 (2019).
17) a) I. Tanabe and Y. Ozaki: Chem. Comm., 50, 2117 (2014). b) I. Tanabe, Y. Yamada, and Y. Ozaki: ChemPhysChem., 17, 516 (2016).
18) a) I. Tanabe, T. Ryoki, and Y. Ozaki: Phys. Chem. Chem. Phys, 16, 7749 (2014). b) I. Tanabe, Y. Kurawaki, Y. Morisawa, and Y. Ozaki: Phys. Chem. Chem. Phys., 18, 22526 (2016).
19) I. Tanabe, A. Suyama, T. Sato, and K. Fukui: Analyst, 143, 2539 (2018).
20) K. B. Bec, Y. Morisawa, K. Kobashi, J. Grabska, I. Tanabe, and Y. Ozaki: J. Phys. Chem. C, 122, 28998 (2018).
21) I. Tanabe, A. Suyama, T. Sato and K. Fukui, Anal. Chem., 91, 3436-3442 (2019).
22) a) Y. Morisawa, N. Higashi, K. Takaba, N. Kariyama, T. Goto, A. Ikehata, and Y. Ozaki: Rev. Sci. Instrum., 83, 073103 (2012). b) T. Goto, Y. Morisawa, N. Higashi, A. Ikehata, and Y. Ozaki: Anal. Chem., 85, 4500 (2013).
23) a) I. Tanabe, Y. Y. Tanaka, T. Ryoki, K. Watari, T. Goto, M. Kikawada, W. Inami, Y. Kawata, and Y. Ozaki: Optics Express, 24, 21886 (2016). b) I. Tanabe, Y. Y. Tanaka, K. Watari, T. Hanulia, T. Goto, W. Inami, Y. Kawata, and Y. Ozaki: Sci. Rep., 7, 5934 (2017).
24) a) N. Higashi, H. Yokota, S. Hirai, and Y. Ozaki, Anal. Chem. 77, 2272 (2005). b) N. Higashi, A. Ikehata, N. Kariyama, and Y. Ozaki, Appl. Spectrosc. 62, 1022 (2008).
25) M. Mitsuoka, H. Shinzawa, Y. Morisawa, N. Kariyama, N. Higashi, M. Tsuboi, and Y. Ozaki, Anal. Sci. 27, 177 (2011).